Diffcalc User Guide (You Engine)¶
Diffcalc: A diffraction condition calculator for diffractometer control
| Author: | Rob Walton |
|---|---|
| Contact: | rob.walton (at) diamond (dot) ac (dot) uk |
| Website: | https://github.com/DiamondLightSource/diffcalc |
See also the quickstart guide at github
Introduction¶
This manual assumes that you are running Diffcalc within OpenGDA or have started it using IPython. It assumes that Diffcalc has been configured for the six circle diffractometer pictured here:
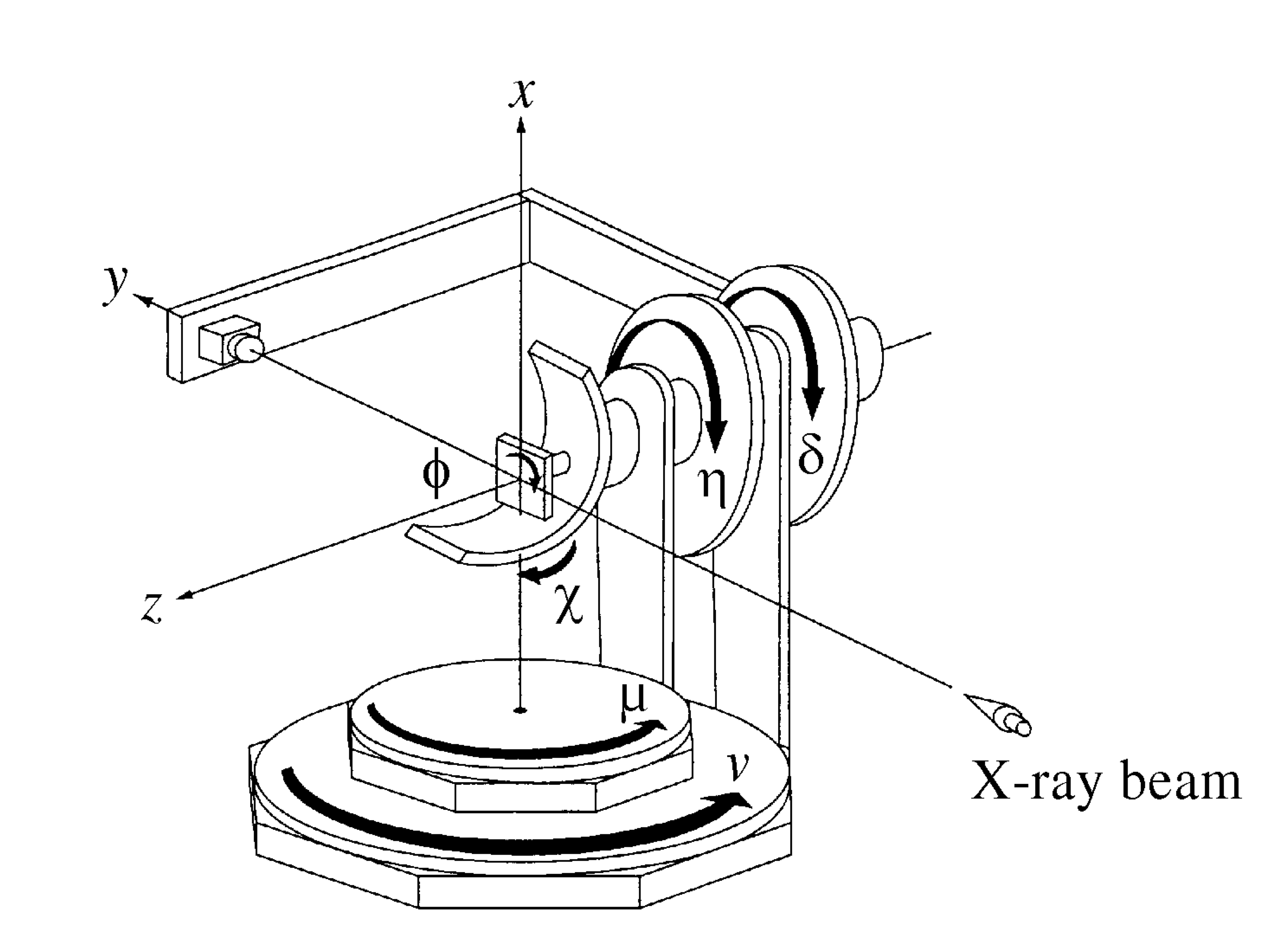
4s + 2d six-circle diffractometer, from H.You (1999)
Your Diffcalc configuration may have been customised for the geometry of your diffractometer and possibly the types of experiment you perform. For example, a five-circle diffractometer might be missing the nu circle above.
The laboratory frame is shown above. With all settings at zero as shown the crystal cartesian frame aligns with the laboratory frame. Therefore a cubic crystal mounted squarely in a way that the U matrix (defined below) is unitary will have h||a||x, k||b||y & l||c||z, crystal and reciprocal-lattice coordinate frames are defined with respect to the beam and to gravity to be (for a cubic crystal):
Overview¶
The following assumes that the diffractometer has been properly levelled, aligned with the beam and zeroed. See the SPEC fourc manual.
Before moving in hkl space you must calculate a UB matrix by specifying the crystal’s lattice parameters (which define the B matrix) and finding two reflections (from which the U matrix defining any mismount can be inferred); and, optionally for surface-diffraction experiments, determine how the surface of the crystal is oriented with respect to the phi axis.
Once a UB matrix has been calculated, the diffractometer may be driven in hkl coordinates. A valid diffractometer setting maps easily into a single hkl value. However for a diffractometer with more than three circles there are excess degrees of freedom when calculating a diffractometer setting from an hkl value. Diffcalc provides modes for using up the excess degrees of freedom.
Diffcalc does not perform scans directly. Instead, Scannables that use diffcalc to map between reciprocal lattice space and real diffractometer settings are scanned using the Gda’s (or minigda’s) generic scan mechanism.
Theory¶
Thanks to Elias Vlieg for sharing his DOS based DIF software that Diffcalc
has borrowed heavily from. The version of Diffcalc described here is based on papers by
pHH. You. [You1999] and Busing & Levy [Busing1967]. (See also the THANKS.txt file.)
Getting Help¶
There are few commands to remember. If a command is called without arguments in some cases Diffcalc will prompt for arguments and provide sensible defaults which can be chosen by pressing enter.
Orientation. The helpub command lists all commands related with crystal
orientation and the reference vector (often used with surfaces). See the
Orientation Commands section at the end of this manual:
>>> help ub
...
HKL movement. The help hkl list all commands related to moving in reciprocal-lattice
space. See the Motion Commands section at the end of this manual:
>>> help hkl
...
Call help on any command. e.g.:
>>> help loadub
loadub (diffcalc command):
loadub 'name' | num -- load an existing ub calculation
Diffcalc’s Scannables¶
To list and show the current positions of your beamline’s scannables
use pos with no arguments:
>>> pos
Results in:
Energy and wavelength scannables:
energy 12.3984
wl: 1.0000
Diffractometer scannables, as a group and in component axes (in the real GDA these have limits):
sixc: mu: 0.0000 delta: 0.0000 gamma: 0.0000 omega: 0.0000 chi: 0.0000 phi: 0.0000
mu: 0.0000
chi: 0.0000
delta: 0.0000
gamma: 0.0000
omega: 0.0000
phi: 0.0000
Dummy counter, which in this example simply counts at 1hit/s:
ct: 0.0000
Hkl scannable, as a group and in component:
hkl: Error: No UB matrix
h: Error: No UB matrix
k: Error: No UB matrix
l: Error: No UB matrix
Parameter scannables, used in some modes, these provide a scannable alternative to the Motion section. Some constrain of these constrain virtual angles:
alpha: ---
beta: ---
naz: ---
psi: ---
qaz: ---
and some constrain physical angles:
phi_con: ---
chi_con: ---
delta_con:---
eta_con: ---
gam_con: ---
mu_con: ---
Crystal orientation¶
Before moving in hkl space you must calculate a UB matrix by specifying the crystal’s lattice parameters (which define the B matrix) and finding two reflections (from which the U matrix can be inferred); and, optionally for surface-diffraction experiments, determine how the surface of the crystal is oriented with respect to the phi axis.
Start a new UB calculation¶
A UB calculation contains the description of the crystal-under-test, any saved reflections, reference angle direction, and a B & UB matrix pair if they have been calculated or manually specified. Starting a new UB calculation will clear all of these.
Before starting a UB-calculation, the ub command used to summarise
the state of the current UB-calculation, will reflect that no
UB-calculation has been started:
>>> ub
<<< No UB calculation started >>>
A new UB-calculation calculation may be started and lattice specified explicitly:
>>> newub 'example'
>>> setlat '1Acube' 1 1 1 90 90 90
or interactively:
>>> newub
calculation name: example
crystal name: 1Acube
crystal system
1) Triclinic
2) Monoclinic
3) Orthorhombic
4) Tetragonal
5) Rhombohedral
6) Hexagonal
7) Cubic
[1]: 7
a[1]: 1
where a is unit cell basis vector in Angstroms for cubic crystal system.
The ub command will show the state of the current UB-calculation
(and the current energy for reference):
>>> ub
UBCALC
name: example
REFERNCE
n_hkl: 1.00000 0.00000 0.00000 <- set
SURFACE NORMAL
n_phi: 0.00000 0.00000 1.00000 <- set
CRYSTAL
name: 1Acube
a, b, c: 1.00000 1.00000 1.00000
90.00000 90.00000 90.00000 Cubic
B matrix: 6.28319 0.00000 0.00000
0.00000 6.28319 0.00000
0.00000 0.00000 6.28319
UB MATRIX
<<< none calculated >>>
REFLECTIONS
<<< none specified >>>
CRYSTAL ORIENTATIONS
<<< none specified >>>
Load a UB calculation¶
To load the last used UB-calculation:
>>> lastub
Loading ub calculation: 'mono-Si'
To load a previous UB-calculation:
>>> listub
UB calculations in: /Users/walton/.diffcalc/i16
0) mono-Si 15 Feb 2017 (22:32)
1) i16-32 13 Feb 2017 (18:32)
>>> loadub 0
Generate a U matrix from two reflections¶
The normal way to calculate a U matrix is to find the position of two
reflections with known hkl values. Diffcalc allows many reflections to be
recorded. After adding first two reflections UB matrix will be calculated
automatically. If there are multiple recorded reflections, the indices or
tags can be provided to calcub command as arguments to calculate UB
matrix from any two given reflections.
Find U matrix from two reflections:
>>> pos wl 1
wl: 1.0000
>>> c2th [0 0 1]
59.99999999999999
>>> pos sixc [0 60 0 30 90 0]
sixc: mu: 0.0000 delta: 60.0000 gam: 0.0000 eta: 30.0000 chi: 90.0000 phi: 0.0000
>>> addref [0 0 1]
>>> pos sixc [0 90 0 45 45 90]
sixc: mu: 0.0000 delta: 90.0000 gam: 0.0000 eta: 45.0000 chi: 45.0000 phi: 90.0000
>>> addref [0 1 1]
Calculating UB matrix.
Check that it looks good:
>>> checkub
ENERGY H K L H_COMP K_COMP L_COMP TAG
1 12.3984 0.00 0.00 1.00 0.0000 0.0000 1.0000
2 12.3984 0.00 1.00 1.00 0.0000 1.0000 1.0000
After adding another reflection we can use the first and the third reflections to recalculate UB matrix:
>>> addref [1 0 1]
>>> calcub 1 3
>>> checkub
ENERGY H K L H_COMP K_COMP L_COMP TAG
1 12.3984 0.00 0.00 1.00 0.0000 0.0000 1.0000
2 12.3984 0.00 1.00 1.00 0.0000 1.0000 1.0000
3 12.3984 1.00 0.00 1.00 1.0000 0.0000 1.0000
Generate a U matrix from one reflection¶
To estimate based on first reflection only:
>>> trialub
resulting U angle: 0.00000 deg
resulting U axis direction: [-1.00000, 0.00000, 0.00000]
Recalculating UB matrix from the first reflection only.
NOTE: A new UB matrix will not be automatically calculated when the orientation reflections are modified.
Edit reflection list¶
Use showref to show the reflection list:
>>> showref
ENERGY H K L MU DELTA GAM ETA CHI PHI TAG
1 12.398 0.00 0.00 1.00 0.0000 60.0000 0.0000 30.0000 90.0000 0.0000
2 12.398 0.00 1.00 1.00 0.0000 90.0000 0.0000 45.0000 45.0000 90.0000
Use swapref to swap reflections:
>>> swapref 1 2
Not calculating UB matrix as it has been manually set. Use 'calcub' to explicitly recalculate it.
Recalculating UB matrix.
Use delref to delete a reflection:
>>> delref 1
Generate a U matrix from two lattice directions¶
Another approach to calculate a U matrix is to provide orientation of two crystal lattice
directions using addorient command after aligning sample in laboratory frame of reference.
The first lattice direction should be aligned along the selected direction in the laboratory frame.
For the purpose of finding azimuthal orientation in U matrix calculation it is sufficient for the
projection of the second lattice direction to be aligned to the given orientation in the laboratory
frame in the plane perpendicular to the first lattice orientation.
Find U matrix from two lattice directions:
>>> addorient [0 0 1] [0 0 1]
>>> addorient [1 0 0] [1 1 0]
Calculating UB matrix.
Calculate a UB matrix¶
Unless a U or UB matrix has been manually specified, a new UB matrix will be calculated after the second reflection has been found, or whenever one of the first two reflections is changed.
Use the command calcub to force the UB matrix to be calculated from the
first two reflections. In case of using lattice orientations instead of reflections,
use command orientub to force the UB matrix to be calculated from the first two orientations.
UB matrix can be calculated from any combination of two reflections and/or orientations
by providing corresponding reflection/orientation tags or numbers as an argument to calcub.
In case of using one reflection and one orientation it is recommended to use tags to avoid
ambiguity.
If you have misidentified a reflection used for the orientation the
resulting UB matrix will be incorrect. Always use the checkub command
to check that the computed reflection indices agree with the estimated values:
>>> checkub
ENERGY H K L H_COMP K_COMP L_COMP TAG
1 12.3984 0.00 1.00 1.00 0.0000 1.0000 1.0000
2 12.3984 0.00 0.00 1.00 0.0000 0.0000 1.0000
Calculate a U matrix from crystal mismount¶
U matrix can be defined from crystal mismount by using a rotation matrix calculated from a provided
mismount angle and axis. setmiscut command defines new U matrix by setting it to a rotation matrix
calculated from the specified angle and axis parameters. addmiscut command applies the calculated
rotation matrix to the existing U matrix, i.e. adds extra mismount to the already existing one:
>>> setmiscut 5 [1 0 0]
n_phi: -0.00000 -0.08716 0.99619
n_hkl: 0.00000 0.00000 1.00000 <- set
normal:
angle: 5.00000
axis: 1.00000 -0.00000 0.00000
Manually specify U matrix¶
Set U matrix manually (pretending sample is squarely mounted):
>>> setu [[1 0 0] [0 1 0] [0 0 1]]
Recalculating UB matrix.
NOTE: A new UB matrix will not be automatically calculated when the orientation reflections are modified.
Refining UB matrix with one reflection¶
UB matrix elements can be refined to match diffractometer settings and crystal orientation experimentally
found for a given reflection with the corresponding reflection indices. refineub command rescales
crystal unit cell dimensions to match with the found scattering angle value and recalculates mismount
parameters to update U matrix:
>>> refineub [1 0 0]
current pos[y]: y
Unit cell scaling factor: 0.99699
Refined crystal lattice:
a, b, c: 0.99699 0.99699 0.99699
90.00000 90.00000 90.00000
Update crystal settings?[y]: y
Warning: the old UB calculation has been cleared.
Use 'calcub' to recalculate with old reflections or
'orientub' to recalculate with old orientations.
Miscut parameters:
angle: 2.90000
axis: -0.00000 1.00000 -0.00000
Apply miscut parameters?[y]: y
n_phi: 0.67043 -0.00000 0.74198
n_hkl: 0.00000 0.00000 1.00000 <- set
normal:
angle: 42.10000
axis: 0.00000 1.00000 0.00000
Calculate UB matrix from multiple reflections¶
Using fitub command UB matrix can be optimised to find best fit for the selected list of reference
reflections . For triclinic crystal system optimal solution is found by solving multivariate linear
regression model, while for the higher symmetry systems it is found by running numerical optimiser:
>>> fitub 1 2 3 4
Fitting crystal lattice parameters...
Fitting orientation matrix...
Refined crystal lattice:
a, b, c: 10.56348 10.56348 10.81364
90.00000 90.00000 90.00000
Update crystal settings?[y]: y
Refined U matrix: 0.94559 -0.32489 0.01762
0.32487 0.94575 0.00437
-0.01809 0.00160 0.99984
Update U matrix?[y]: y
...
...
...
REFLECTIONS
ENERGY H K L PHI CHI ETA MU DELTA GAM TAG
1 8.000 0.00 0.00 8.00 -26.3000 89.0000 17.3034 0.0005 33.3569 -0.0042 None
2 8.000 4.00 4.00 8.00 62.4273 53.4451 45.2680 0.0000 90.0825 0.0000 None
3 8.000 0.00 0.00 8.00 13.3485 89.0097 35.0408 0.0000 69.9326 0.0000 None
4 8.000 4.00 4.00 8.00 63.2008 53.4096 44.9007 0.0000 90.1107 0.0000 None
Set the reference vector¶
The reference vector can be used to define azimuthal direction within the crystal
with which we want to orient the incident or diffracted beam. Orientation of the reference vector
w.r.t the incident and diffracted beam is indicated using alpha and beta angles.
By default the reference vector is set parallel to the theta axis. That is, along the x-axis of the laboratory coordinate frame.
The ub command shows the current reference vector at the top its report
(or it can be shown by calling setnphi or setnhkl with no args):
>>> ub
...
REFERNCE
n_phi: 1.00000 0.00000 0.00000
n_hkl: 1.00000 0.00000 0.00000 <- set
...
The <- set label here indicates that the reference vector is set in the reciprocal
lattice space. In this case, therefore, its direction in the laboratory coordinate
frame is inferred from the UB matrix.
To set the reference vector in the phi coordinate frame use:
>>> setnphi [1 0 0]
...
To set the reference vector in the crystal’s reciprocal lattice space use:
>>> setnhkl [1 0 0]
...
Set the surface normal vector¶
The orientation of the sample surface can be set using the surface normal vector defined either in
laboratory coordinate system or reciprocal space. Orientation of the surface normal vector
w.r.t the incident and diffracted beam is indicated using betain and betaout angles.
By default the surface normal vector is set parallel to the phi axis. That is, along the z-axis of the laboratory coordinate frame.
The ub command shows the current surface normal vector at the top its report
(or it can be shown by calling surfnphi or surfnhkl with no args):
>>> ub
...
SURFACE NORMAL
n_phi: 0.00000 0.00000 1.00000 <- set
n_hkl: 0.00000 0.00000 1.00000
...
The <- set label here indicates that the surface normal vector is set in the laboratory
coordinate frame. In this case, therefore, its direction in the crystal’s
reciprocal lattice space is inferred from the UB matrix.
To set the surface normal vector in the phi coordinate frame use:
>>> surfnphi [0 0 1]
...
To set the surface normal vector in the crystal’s reciprocal lattice space use:
>>> surfnhkl [0 0 1]
...
Motion¶
Once a UB matrix has been calculated, the diffractometer may be driven in hkl coordinates. A given diffractometer setting maps easily into a single hkl value. However for a diffractometer with more than three circles there are excess degrees of freedom when calculating a diffractometer setting from an hkl value. Diffcalc provides many for using up the excess degrees of freedom.
By default Diffcalc selects no mode.
Constraining solutions for moving in hkl space¶
To get help and see current constraints:
>>> help con
...
>>> con
DET REF SAMP
----------- ----------- -----------
delta a_eq_b mu
gam alpha eta
qaz beta chi
naz psi phi
bin_eq_bout mu_is_gam
betain bisect
betaout omega
! 3 more constraints required
Type 'help con' for instructions
Three constraints can be given: zero or one from the DET and REF columns and the
remainder from the SAMP column. Not all combinations are currently available.
Use help con to see a summary if you run into troubles.
To configure four-circle vertical scattering:
>>> con gam 0 mu 0 a_eq_b
gam : 0.0000
a_eq_b
mu : 0.0000
In the following the scattering plane is defined as the plane including the scattering vector, or momentum transfer vector, and the incident beam.
DETECTOR COLUMN:
- delta - physical delta setting (vertical detector motion) del=0 is equivalent to qaz=0
- gam - physical gamma setting (horizontal detector motion) gam=0 is equivalent to qaz=90
- qaz - azimuthal rotation of scattering vector (about the beam, from horizontal)
- naz - azimuthal rotation of reference vector (about the beam, from horizontal)
REFERENCE COLUMN:
- alpha - incident angle to reference vector
- beta - exit angle from reference vector
- psi - azimuthal rotation about scattering vector of reference vector (from scattering plane)
- a_eq_b - bisecting mode with alpha=beta. Equivalent to psi=90
- betain - incident angle to sample surface
- betaout - exit angle from sample surface
- bin_eq_bout - bisecting mode with betain=betaout
SAMPLE COLUMN:
- mu, eta, chi & phi - physical settings
- mu_is_gam - force mu to follow gamma (results in a 5-circle geometry)
- bisect - bisecting mode with scattering vector in chi-circle plane
- omega - bisecting mode with omega angle between scattering vector and chi-circle plane
Diffcalc will report two other (un-constrainable) virtual angles:
- theta - half of 2theta, the angle through the diffracted beam bends
- tau - longitude of reference vector from scattering vector (in scattering plane)
Example constraint modes¶
There is sometimes more than one way to get the same effect.
Vertical four-circle mode:
>>> con gam 0 mu 0 a_eq_b # or equivalently:
>>> con qaz 90 mu 0 a_eq_b
>>> con alpha 1 # replaces a_eq_b
Horizontal four-circle mode:
>>> con del 0 eta 0 alpha 1 # or equivalently:
>>> con qaz 0 mu 0 alpha 1
Surface vertical mode:
>>> con naz 90 mu 0 betain 1
Surface horizontal mode:
>>> con naz 0 eta 0 betain 1
Z-axis mode (surface horizontal):
>>> con chi (-sigma) phi (-tau) betain 1
where sigma and tau are the offsets required in chi and phi to bring the surface normal parallel to eta. betain will determine mu directly leaving eta to orient the planes. Or:
>>> con naz 0 phi 0 betain 1 # or any another sample angle
Z-axis mode (surface vertical):
>>> con naz 0 phi 0 betain 1 # or any another sample angle
Changing constrained values¶
Once constraints are chosen constrained values may be changed directly:
>>> con mu 10
gam : 0.0000
a_eq_b
mu : 10.0000
or via the associated scannable:
>>> pos mu_con 10
mu_con: 10.00000
Configuring limits and cuts¶
Diffcalc uses motor limits set in GDA when used from GDA client running on a beamline. The standalone console version maintains its own limits on axes. These limits will be used when choosing solutions. If more than one detector solution exists Diffcalc will ask you to reduce the the limits until there is only one. However if more than one solution for the sample settings is available it will choose one that is closest to the current diffractometer orientation.
Use the hardware command to see the current limits and cuts:
>>> hardware
mu (cut: -180.0)
delta (cut: -180.0)
gam (cut: -180.0)
eta (cut: -180.0)
chi (cut: -180.0)
phi (cut: 0.0)
Note: When auto sector/transforms are used,
cuts are applied before checking limits.
To set the limits in standalone Diffcalc session:
>>> setmin delta -1
>>> setmax delta 145
To set a cut:
>>> setcut phi -180
This causes requests to move phi to be between the configured -180 and +360 degress above this. i.e. it might dive to -10 degrees rather than 350.
Moving in hkl space¶
Configure a mode, e.g. four-circle vertical:
>>> con gam 0 mu 0 a_eq_b
gam : 0.0000
a_eq_b
mu : 0.0000
Simulate moving to a reflection:
>>> sim hkl [0 1 1]
sixc would move to:
mu : 0.0000
delta : 90.0000
gam : 0.0000
eta : 45.0000
chi : 45.0000
phi : 90.0000
alpha : 45.0000
beta : 45.0000
betain : 30.0000
betaout : 30.0000
naz : 35.2644
psi : 90.0000
qaz : 90.0000
tau : 45.0000
theta : 45.0000
ttheta : 90.0000
Move to reflection:
>>> pos hkl [0 1 1]
hkl: h: 0.00000 k: 1.00000 l: 1.00000
>>> pos sixc
sixc: mu: 0.0000 delta: 90.0000 gam: 0.0000 eta: 45.0000 chi: 45.0000 phi: 90.0000
Simulate moving to a location:
>>> pos sixc [0 60 0 30 90 0]
sixc: mu: 0.0000 delta: 60.0000 gam: 0.0000 eta: 30.0000 chi: 90.0000 phi: 0.0000
Scanning in hkl space¶
All scans described below use the same generic scanning mechanism provided by the GDA system or by minigda. Here are some examples.
Fixed hkl scans¶
In a ‘fixed hkl scan’ something (such as energy or Bin) is scanned, and at each step hkl is ‘moved’ to keep the sample and detector aligned. Also plonk the diffractometer scannable (sixc) on there with no destination to monitor what is actually happening and then throw on a detector (ct) with an exposure time if appropriate:
>>> #scan scannable_name start stop step [scannable_name [pos or time]]..
>>> scan en 9 11 .5 hkl [1 0 0] sixc ct 1
>>> scan en 9 11 .5 hklverbose [1 0 0] sixc ct 1
>>> scan betain 4 5 .2 hkl [1 0 0] sixc ct 1
>>> scan alpha_par 0 10 2 hkl [1 0 0] sixc ct 1
Scanning hkl¶
Hkl, or one component, may also be scanned directly:
>>> scan h .8 1.2 .1 hklverbose sixc ct 1
At each step, this will read the current hkl position, modify the h component and then move to the resulting vector. There is a danger that with this method k and l may drift. To get around this the start, stop and step values may also be specified as vectors. So for example:
>>> scan hkl [1 0 0] [1 .3 0] [1 0.1 0] ct1
is equivilant to:
>>> pos hkl [1 0 0]
>>> scan k 0 .3 .1 ct1
but will not suffer from drifting. This method also allows scans along any direction in hkl space to be performed.
Multidimension scans¶
Two and three dimensional scans:
>>> scan en 9 11 .5 h .9 1.1 .2 hklverbose sixc ct 1
>>> scan h 1 3 1 k 1 3 1 l 1 3 1 hkl ct 1
Commands¶
Orientation Commands¶
| STATE | |
| – newub {‘name’} | start a new ub calculation name |
| – loadub ‘name’ | num | load an existing ub calculation |
| – lastub | load the last used ub calculation |
| – listub | list the ub calculations available to load |
| – rmub ‘name’ | num | remove existing ub calculation |
| – saveubas ‘name’ | save the ub calculation with a new name |
| LATTICE | |
| – setlat | interactively enter lattice parameters (Angstroms and Deg) |
| – setlat name a | assumes cubic |
| – setlat name a b | assumes tetragonal |
| – setlat name a b c | assumes ortho |
| – setlat name a b c gamma | assumes mon/hex with gam not equal to 90 |
| – setlat name a b c alpha beta gamma | arbitrary |
| – c2th [h k l] | calculate two-theta angle for reflection |
| – hklangle [h1 k1 l1] [h2 k2 l2] | calculate angle between [h1 k1 l1] and [h2 k2 l2] crystal planes |
| REFERENCE | |
| – setnphi {[x y z]} | sets or displays n_phi reference |
| – setnhkl {[h k l]} | sets or displays n_hkl reference |
| SURFACE NORMAL | |
| – surfnphi {[x y z]} | sets or displays surface normal vector in lab space |
| – surfnhkl {[h k l]} | sets or displays surface normal vector in reciprocal space |
| REFLECTIONS | |
| – showref | shows full reflection list |
| – addref | add reflection interactively |
| – addref [h k l] {‘tag’} | add reflection with current position and energy |
| – addref [h k l] (p1, .., pN) energy {‘tag’} | add arbitrary reflection |
| – editref num | interactively edit a reflection |
| – delref num | deletes a reflection (numbered from 1) |
| – clearref | deletes all the reflections |
| – swapref | swaps first two reflections used for calculating U matrix |
| – swapref num1 num2 | swaps two reflections (numbered from 1) |
| CRYSTAL ORIENTATIONS | |
| – showorient | shows full list of crystal orientations |
| – addorient | add crystal orientation interactively |
| – addorient [h k l] [x y z] {‘tag’} | add crystal orientation in laboratory frame |
| – editorient num | interactively edit a crystal orientation |
| – delorient num | deletes a crystal orientation (numbered from 1) |
| – clearorient | deletes all the crystal orientations |
| – swaporient | swaps first two crystal orientations used for calculating U matrix |
| – swaporient num1 num2 | swaps two crystal orientations (numbered from 1) |
| UB MATRIX | |
| – fitub ref1 ref2 ref3 .. | fit UB matrix to match list of provided reference reflections |
| – checkub | show calculated and entered hkl values for reflections |
| – setu {[[..][..][..]]} | manually set u matrix |
| – setub {[[..][..][..]]} | manually set ub matrix |
| – calcub | (re)calculate u matrix from ref1 and ref2 |
| – calcub idx1 idx2 | (re)calculate U matrix from reflections and/or orientations referred by indices and/or tags idx1 and idx2 |
| – orientub | (re)calculate U matrix from reflections and/or orientations |
| – orientub idx1 idx2 | (re)calculate U matrix from the first two orientations referred by indices and/or tags idx1 and idx2 |
| – trialub | (re)calculate U matrix from the first reflection (check carefully) |
| – trialub idx1 | (re)calculate U matrix from reflection with index or tag idx only (check carefully) |
| – refineub {[h k l]} {pos} | refine unit cell dimensions and U matrix to match diffractometer angles for a given hkl value |
| – addmiscut angle {[x y z]} | apply miscut to U matrix using a specified miscut angle in degrees and a rotation axis (default: [0 1 0]) |
| – setmiscut angle {[x y z]} | manually set U matrix using a specified miscut angle in degrees and a rotation axis (default: [0 1 0]) |
Motion commands¶
| CONSTRAINTS | |
| – con | list available constraints and values |
| – con <name> {val} | constrains and optionally sets one constraint |
| – con <name> {val} <name> {val} <name> {val} | clears and then fully constrains |
| – uncon <name> | remove constraint |
| HKL | |
| – allhkl [h k l] | print all hkl solutions ignoring limits |
| HARDWARE | |
| – hardware | show diffcalc limits and cuts |
| – setcut {name {val}} | sets cut angle |
| – setmin {axis {val}} | set lower limits used by auto sector code (None to clear) |
| – setmax {name {val}} | sets upper limits used by auto sector code (None to clear) |
| MOTION | |
| – sim hkl scn | simulates moving scannable (not all) |
| – sixc | show Eularian position |
| – pos sixc [mu, delta, gam, eta, chi, phi] | move to Eularian position(None holds an axis still) |
| – sim sixc [mu, delta, gam, eta, chi, phi] | simulate move to Eulerian positionsixc |
| – hkl | show hkl position |
| – pos hkl [h k l] | move to hkl position |
| – pos {h | k | l} val | move h, k or l to val |
| – sim hkl [h k l] | simulate move to hkl position |
Good luck — RobW
References¶
| [You1999] | H. You. Angle calculations for a ‘4S+2D’ six-circle diffractometer. J. Appl. Cryst. (1999). 32, 614-623. (pdf link). |
| [Busing1967] | W. R. Busing and H. A. Levy. Angle calculations for 3- and 4-circle X-ray and neutron diffractometers. Acta Cryst. (1967). 22, 457-464. (pdf link). |